Document
Background
This section provides the basic knowledge of CRISPR-Cas9.
CRISPR
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) is an acronym for DNA loci that contain multiple, short, direct repetitions of base sequences. Each repetition contains a series of base pairs followed by the same or a similar series in reverse and then by 30 or so base pairs known as "spacer DNA". CRISPRs are found in the genomes of approximately 40% of sequenced bacteria and 90% of sequenced archaea (Grissa et al., 2007). CRISPRs are often associated with cas genes which code for proteins that perform various functions related to CRISPRs. The CRISPR-Cas system functions as a prokaryotic immune system, in that it confers resistance to exogenous genetic elements such as plasmids and phages and provides a form of acquired immunity. CRISPR spacers recognize and silence exogenous genetic elements in a manner analogous to RNAi in eukaryotic organisms (Marraffini and Sontheimer, 2010) .
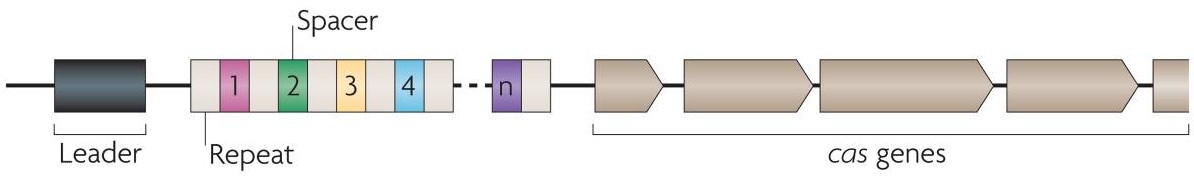
CRISPR-Cas9 system in application
As CRISPR-Cas9 immune system takes aim at specific DNA sequences, it has been used for a novel technique of gene editing that even works in eukaryotes. A single nuclease Cas9 is guided to the target DNA by a guide RNA (gRNA) which contains a sequence that matches the sequence to be cleaved. Efficient cleavage also requires the presence of 5’-NGG-3’ Protospacer Adjacent Motif (PAM) sequences following the guide sequence. RNA-guided Cas9 creates site-specific double-stranded DNA breaks, which are then repaired by either non-homologous end joining or homologous recombination. (Jinek et al., 2012).
In January 2013, four research teams reported harnessing the system to target the destruction of specific genes in human cells. And in the following 8 months, various groups have used it to delete, add, activate, or suppress targeted genes in human cells, mice, rats, zebrafish, bacteria, fruit flies, yeast, nematodes, and crops, demonstrating broad utility for the technique (Pennisi 2013).
Optimal gRNA design
Hsu et al. characterize Cas9 targeting specificity in human cells to inform the selection of target sites and avoid off-target effects. In which, they find that Cas9 tolerates mismatches between guide RNA and target DNA at different positions in sequence-dependent manner, sensitive to the number, position and distribution of mismatches (Hsu et al., 2013). We provide Optimized CRISPR-Plant Design Tool to guide the selection of optimized gRNA for target sequences by minimizing the potential off-target effects.
gRNA Design Guide
This paragraph describes how to use the Optimized CRISPR-Plant Design tool.
Quick Guide - optimal gRNA design overview
The following list provides a swift overview of the steps involved in gRNA design:
- Please read the step by step guide to prevent frustration and to ensure optimal results.
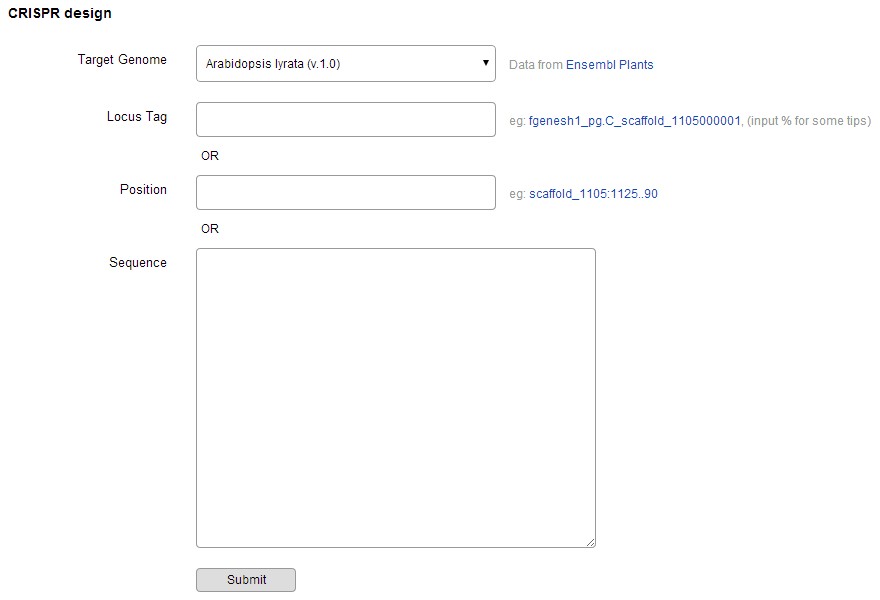
- Select a genome you are studying, input gene locus, chromosome position, or DNA sequence.
- If your sequence could not be found in the blast search of your selected genome, the work is not allowed to proceed!
- After submission, 3 steps are taken automatically. The gene model of the target gene are showed in graphic and the gRNA candidates are sorted according to its off-target potential, the gRNA at the top of the list are expected to work better in most cases.
- Mouseover each gRNA candidate to see potential off-target site in the genome. Besides, restriction enzyme cutting sites are indicated on every gRNA.
- Choose a better choice among the results according to your needs.
Procedure - optimal gRNA design step by step
The following displays a detailed procedure for optimal gRNA design.
- Input sequences
- Design optimized gRNA
- gRNA selection
Optimized CRISPR-P design tool supports many plant species. When a user selects a plant species and submits a DNA sequence (or gene locus, chromosome position), the input sequence is aligned to the target plant genome by using BLASTN, the best BLASTN hit with the same length, no mismatch and no gap is selected. Sequence in the range of 23 ~ 5000 bp is suggested to input. If the input sequence is not in the BLASTN results of the target genome, the task will be stopped.
Click "submit", the calculating will be very fast and longer sequence will need more time, in which, 3 steps are taken sequentially.
Step 1, the target sequence is mapped to its genome, and all possible gRNA are screened out and showed in graphic gene model (20 nucleotides followed by a PAM sequence: NGG). Annotation informations of the target sequence are also showed below the gene model.
Step 2, every gRNA sequences will be scanned for possible off-target matches throughout the selected genome. The off-target sites are scored according to their interference potential, and top 20 sites with their mismatch (MMs) number to target sequence are listed for every gRNA (red color highlight the mismatch sites). Then, an aggregate score are calculated for each gRNA, and we colored the presumably best ones in red (score > 50), followed by intermediate ones in green (20 < score < 50), and the remaining ones in grey at the bottom of the list. gRNA are also can be highlighted in the graphic gene model when mouseover in the list. Please read our "Selection criteria" for scoring information.
Step 3, informations of potential off-target sites for every gRNA are been displayed. In which, commonly used restriction enzyme cutting site are displayed, and gene names will be showed if the off-target site is placed in a gene region.

One can choose a better choice among the possible gRNAs according to its specificity, location (gene exon, intron or promoter), or preferred restriction enzyme cutting site.
(Some features require your web browsers with supporting HTML5)Selection criteria
This section introduces the scoring criteria that we use in optimal gRNA design.
Off-target sites searching for every gRNA
When screening potential off-target sites for every gRNA, “batmis” software is used to perform the alignment between every gRNA and the target genome. At most 4 mismatches are allowed for the first 20nt. And for PAM sequence, 5’-NAG are also considered as off-target sites as it got one-fifth efficiency for targets with 5’-NGG PAMs (Hsu et al, 2013).
Scoring for off-target potential
The algorithm used to score single offtargets is:
Within the first term, e runs over the mismatch positions between guide and offtarget, with M:
M = [0,0,0.014,0,0,0.395,0.317,0,0.389,0.079,0.445,0.508,0.613,0.851,0.732,0.828,0.615,0.804,0.685,0.583]
representing the experimentally-determined effect of mismatch position on targeting. (The algorithm is cited from Hsu et al, 2013.)
Aggregate scoring for all possible gRNAs
Once individual hits have been scored, each guide is assigned a score:
sgRNAs are colored according to a broad categorization of guide quality, which taken into account with the presence or absence of marked genes in high-scoring offtargets indicate the relative (un)favorability of using a particular guide for specific targeting in the query region. (The algorithm is cited from Hsu et al, 2013.)
Contact
Project Leader: Lingling Chen
Any question contact us